
Las enfermedades por almacenamiento de lípidos, o lipidosis, son un grupo de trastornos metabólicos heredados en los cuales cantidades perjudiciales de materiales grasos llamados lípidos se acumulan en algunas de las células y tejidos del cuerpo. Las personas con estos trastornos no producen suficiente de una de las enzimas necesarias para metabolizar los lípidos o producen enzimas que no funcionan adecuadamente. Con el tiempo, este almacenamiento excesivo de grasas puede causar daño tisular y celular permanente, particularmente en el cerebro, el sistema nervioso periférico, el hígado, el bazo y la médula ósea.
Los lípidos son sustancias parecidas a las grasas que son partes importantes de las membranas encontradas dentro y entre las células y en la vaina de mielina que recubre y protege los nervios. Los lípidos incluyen a los aceites, ácidos grasos, ceras, esteroides (como el colesterol y el estrógeno), y otros compuestos vinculados.
Estos materiales grasos se almacenan naturalmente en las células, órganos y tejidos del cuerpo. Diminutos cuerpos dentro de las células llamados lisosomas habitualmente convierten o metabolizan a los lípidos y las proteínas a componentes más pequeños para proporcionar energía para el cuerpo. Los trastornos que almacenan este material intracelular se llaman enfermedades por almacenamiento de lisosomas. Además de las enfermedades por almacenamiento de lípidos, otras enfermedades por almacenamiento de lisosomas son las mucolipidosis, en las cuales cantidades excesivas de lípidos y moléculas de azúcar se almacenan en los tejidos y las células, y las mucopolisacaridosis, en las cuales se almacenan cantidades excesivas de moléculas de azúcar.
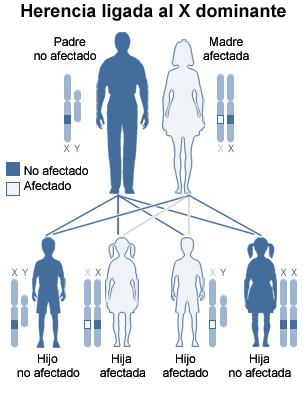
¿Las enfermedades por almacenamiento de lípidos se heredan de uno o ambos padres que transportan el gen defectuoso que regula una proteína particular en una clase de células del cuerpo.
El diagnóstico se hace por el examen clínico, biopsia, pruebas genéticas, análisis molecular de las células o tejido para identificar los trastornos metabólicos heredados, y ensayos enzimáticos (para evaluar una variedad de células o líquidos corporales en cultivo para detectar deficiencias enzimáticas). En algunas formas del trastorno, un análisis de orina puede identificar la presencia de material almacenado. Algunas pruebas también pueden determinar si una persona transporta el gen defectuoso que puede trasmitirse a sus hijos. Este proceso se conoce como genotipificación.
La enfermedad de Fabry, también conocida como deficiencia de la alfa-galactosidasa-A, causa una acumulación de material graso en el sistema nervioso autónomo, los ojos, los riñones, y el sistema cardiovascular. La enfermedad de Fabry es la única enfermedad por almacenamiento de lípidos ligado a X. Los hombres están principalmente afectados aunque es común una forma más leve en las mujeres, algunas de las cuales tienen manifestaciones graves similares a las vistas en los hombres afectados. Generalmente el inicio de los síntomas es durante la niñez o la adolescencia.
Los síntomas neurológicos incluyen dolor quemante en los brazos y las piernas, que empeora en agua caliente o luego de hacer ejercicio, y la acumulación de material de exceso en las capas transparentes de la cornea (dando como resultado una nubosidad pero ningún cambio en la visión). Los depósitos grasos en las paredes de los vasos sanguíneos pueden deteriorar la circulación, colocando al paciente en riesgo de tener un accidente cerebrovascular o un ataque cardíaco.
Otros síntomas incluyen el agrandamiento cardíaco, deterioro progresivo de los riñones que lleva a insuficiencia renal, dificultades gastrointestinales, disminución del sudor y fiebre. Pueden desarrollarse angioqueratomas (manchas elevadas pequeñas, no cancerosas, de color púrpura-rojizo en la piel) en la parte inferior del tronco y volverse más numerosas con la edad.
Los pacientes con enfermedad de Fabry a menudo mueren prematuramente de complicaciones de enfermedad cardíaca, insuficiencia renal o accidente cerebrovascular. A menudo se recetan medicamentos como la fenitoína y la carbamazepina para tratar el dolor que acompaña a la enfermedad de Fabry. Metoclopramida o Lipisorb (un complemento nutricional) puede aliviar el malestar gastrointestinal que se produce a menudo en los pacientes con Fabry, y algunos individuos pueden necesitar un trasplante o diálisis renal.
Experimentos recientes indican que el reemplazo enzimático puede reducir el almacenamiento, aliviar el dolor, y mejorar la función orgánica de los pacientes con la enfermedad de Fabry.